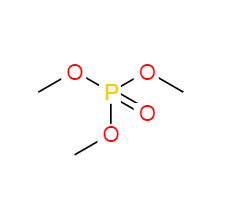
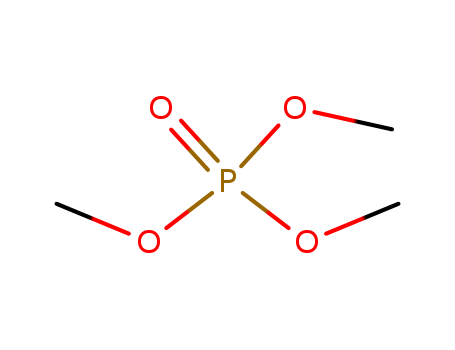
Trimethyl phosphate literature
Complexation and Chemisorption of Trimethylphosphine on Ni Zeolites
Schoonheydt, Robert A.,Wouwe, Dirk Van,Leeman, Hugo
, p. 2519 - 2530 (1980)
After room temperature saturation of dehydrated NiY with trimethylphosphine two complexes are formed in the supercages.They are identified and quantified by reflectance spectroscopy as 0.92+ per unit cell and 6-8<(Ol)3-Ni-PMe3>2+ per unit cell (Ol is a lattice oxygen).The former is diamagnetic and trigonal bipyramidal, the latter is paramagnetic and compressed tetrahedral. 2+ is only stable in excess PMe3, while the mono-phosphine complex is stable to ca. 383 K in vacuo.On NiX only the paramagnetic compressed tetrahedral complex is formed.The ligand field parameters of this complex were calculated.Chemisorption on lattice and extralattice oxygens gives strongly held O=PMe3, O=P(OMe)3 and a range of decomposition products such as CO, CO2, H2O, hydrocarbons and oxygenated P on the surface.These products were qualitatively identified by i.r. and mass spectrometry.
Solvent isotope effects as a probe of general catalysis and solvation in phosphoryl transfer
Bryan,Schowen,Schowen
, p. 931 - 938 (1996)
Phosphoryl transfer to methanol from PNNN, PMNN, and PMMN exhibits general base catalysis by acetate ion but no detectable catalysis by acetic acid. For PNNN, acetate catalysis produces normal solvent isotope effects that arise from a one-proton catalytic bridge in the transition state. The proton inventory for the least reactive substrate PMMN is suggestive of transition-state stabilization, while the proton inventory for the most reactive substrate PNNN suggests only generalized transition-state solvation. Furthermore, the proton inventory for PMNN suggests an intermediate situation. The data are consistent with a model in which transition states with exterior concentrations of charge favor stabilization of the charge by isotope-fractionating one-proton bridges, while transition states with distributed charge favor stabilization of the charge by many distributed sites.
Catalysis of the oxidation of triphenylphosphine and of trimethyl phosphite by hydrogen peroxide in the presence of Fe(III) compounds
Barton, Derek H.R.,Hill, David R.,Hu, Bin
, p. 1711 - 1712 (1997)
The oxidation of triphenylphosphine to the corresponding oxide in pyridine is faster in the presence of Fe(III) compounds, especially FeCl3. Even greater effects are seen for the oxidation of trimethyl phosphite.
Preparation and reactivity of half-sandwich dioxygen complexes of ruthenium
Albertin, Gabriele,Antoniutti, Stefano,Bortoluzzi, Marco,Castro, Jesús,Ferraro, Valentina
, p. 9173 - 9184 (2018)
Dioxygen complexes [Ru(η5-C5Me5)(η2-O2){P(OEt)3}2]BPh4 (1) and [Ru(η5-C5Me5)(η2-O2)(PPh3){P(OR)3}]BPh4 (2, 3) [R = Me (2), Et (3)] were prepared by allowing chloro-complexes RuCl(η5-C5Me5)[P(OEt)3]2 and RuCl(η5-C5Me5)(PPh3)[P(OR)3] to react with air (1 atm) in the presence of NaBPh4. Substitution of the η2-O2 in 1-3 by alkenes [CH2CH2, CHCHCO(O)CO] and terminal alkynes (PhCCH) afforded [Ru(η5-C5Me5)(η2-CH2CH2){P(OEt)3}L]BPh4 (4) [L = P(OEt)3 (a), PPh3 (b)], [Ru(η5-C5Me5){η2-CHCHCO(O)CO}{P(OEt)3}2]BPh4 (5) and [Ru(η5-C5Me5){CC(H)Ph}{P(OEt)3}2]BPh4 (6) derivatives. Protonation of dioxygen complexes 1-3 with triflic acid yielded phosphate complexes [Ru(κ1-OTf)(η5-C5Me5){P(O)(OEt)3}2] (7) and [Ru(κ1-OTf)(η5-C5Me5){P(O)Ph3}{P(O)(OMe)3}] (8). A reaction path for the formation of complexes 7 and 8 is proposed by DFT studies. Besides phosphate complex 7, protonation of 1 under a CH2CH2 atmosphere (1 atm) afforded acetic acid. Treatment of complexes 7 and 8 with tBuNC afforded the tris(isocyanide) derivative [Ru(η5-C5Me5)(tBuNC)3]BPh4 (9). The complexes were characterised spectroscopically (IR and NMR) and by X-ray crystal structure determination of 1, 2 and 3.
Photooxidation of Trimethyl Phosphite in Nitrogen, Oxygen, and para-Hydrogen Matrixes at Low Temperatures
Ramanathan,Sundararajan,Gopi,Sankaran
, p. 2121 - 2131 (2017)
(Chemical Equation Presented) Trimethyl phosphite (TMPhite) was photooxidized to trimethyl phosphate (TMP) in N2, O2, and para-H2 matrixes at low temperatures to correlate the conformational landscape of these two molecules. The photooxidation produced the trans (TGG)-rich conformer with respect to the ground state gauche (GGG) conformer of TMP in N2 and O2 matrixes, which has diverged from the conformational composition of freshly deposited pure TMP in the low-temperature matrixes. The enrichment of the trans conformer in preference to the gauche conformer of TMP during photooxidation is due to the TMPhite precursor, which exists exclusively in the trans conformer. Interestingly, whereas the photooxidized TMP molecule suffers site effects possibly due to the local asymmetry in N2 and O2 matrixes, in the para-H2 matrix owing to the quantum crystal nature the site effects were observed to be self-repaired.
-
Cohen
, p. 3491 (1965)
-
Photoinduced single electron transfer activation of organophosphines: Nucleophilic trapping of phosphine radical cation
Pandey, Ganesh,Pooranchand, Dinah,Bhalerao
, p. 1745 - 1752 (1991)
Photophysical studies show that organophosphines (1-4) form charge transfer stabilized exciplex with excited singlet 1DCN*. One electron oxidation of phosphines to corresponding radical cation via phosphine... 1DCN* electron donor, acceptor pair dissociation is reported. Phosphine radical cations are lound to react readily with moisture to give phosphine oxides.
Rates and Mechanisms of Hydrolysis of Esters of Phosphorous Acid
Westheimer, F. H.,Huang, Shaw,Covitz, Frank
, p. 181 - 185 (1988)
Trialkyl phosphites hydrolyze in alkali several hudred times as fast as do the corresponding phosphates; in acid, however, the phosphite hydrolyzes 1E12 times asa fast as the phosphate.Dialkyl phosphites (i.e., dialkyl hydrogen phosphonates) hydrolyze in alkali more than 1E5 times as fast as do the corresponding triesters, presumably by way of metaphosphites as intermediates.The mechanisms of these various processes are discussed.The date are of interest in connection with the phosphite syntheses of DNA.
Terminal Titanyl Complexes of Tri- and Tetrametaphosphate: Synthesis, Structures, and Reactivity with Hydrogen Peroxide
Stauber, Julia M.,Cummins, Christopher C.
, p. 3022 - 3029 (2017)
The synthesis and characterization of tri- and tetrametaphosphate titanium(IV) oxo and peroxo complexes is described. Addition of 0.5 equiv of [OTi(acac)2]2 to dihydrogen tetrametaphosphate ([P4O12H2]2-) and monohydrogen trimetaphosphate ([P3O9H]2-) provided a bis(μ2,κ2,κ2) tetrametaphosphate titanyl dimer, [OTiP4O12]24- (1; 70% yield), and a trimetaphosphate titanyl acetylacetonate complex, [OTiP3O9(acac)]2- (2; 59% yield). Both 1 and 2 have been structurally characterized, crystallizing in the triclinic P1? and monoclinic P21 space groups, respectively. These complexes contain TiO units with distances of 1.624(7) and 1.644(2) ?, respectively, and represent rare examples of structurally characterized terminal titanyls within an all-oxygen coordination environment. Complexes 1 and 2 react with hydrogen peroxide to produce the corresponding peroxotitanium(IV) metaphosphate complexes [O2TiP4O12]24-(3; 61% yield) and [O2TiP3O9(acac)]2- (4; 65% yield), respectively. Both 3 and 4 have been characterized by single-crystal X-ray diffraction studies, and their solid-state structures are presented. Complex 3 functions as an oxygen atom transfer (OAT) reagent capable of oxidizing phosphorus(III) compounds (P(OMe)3, PPh3) and SMe2 at ambient temperature to result in the corresponding organic oxide with regeneration of dimer 1.
Oxidizing O,O,O-trimethyl phosphorothioate with hydrogen peroxide or ozone
Chen, Xinzhi,Li, Guihua,Qian, Chao
, p. 645 - 649 (2012)
Oxidization of O,O,O-trimethyl phosphorothioate to the corresponding oxo (P=O) compound is accomplished under mild conditions with hydrogen peroxide or ozone. Urea hydroperoxide was found to be a serviceable solid-state source of hydrogen peroxide; it was a better oxidizing agent than 30% hydrogen peroxide and resulted in better yield. It was also proved that ozone was the most effective oxidizer, with formation of purer products in higher yield (90.1%) than with hydrogen peroxide. Springer Science+Business Media B.V. 2011.
METHOD FOR PRODUCING PHOSPHOESTER COMPOUND
-
Paragraph 0023; 0026-0028, (2021/09/27)
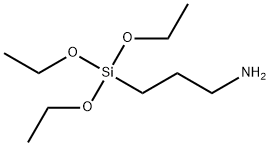
PROBLEM TO BE SOLVED: To provide a method whereby, a phosphate compound selected from the group consisting of orthophosphoric acid, phosphonic acid, phosphinic acid, and anhydrides of them is used as raw material and, by one stage reaction, a corresponding phosphoester compound is produced. SOLUTION: To an aqueous solution of a phosphate compound, added is an organic silane or siloxane compound having an alkoxy group or an aryloxy group, and the mixture is subjected to a heating reaction, thereby producing a corresponding phosphoester compound without requiring a catalyst. SELECTED DRAWING: None COPYRIGHT: (C)2021,JPOandINPIT
Method for compounding organphosphorus by using black phosphorus
-
Paragraph 0054; 0055; 0056; 0060, (2018/10/19)
The invention discloses a method for compounding organphosphorus by using black phosphorus. The method takes the black phosphorus as a reaction raw material, safely and efficiently compounds organic phosphate, sulpho-phosphite ester, amino phosphite ester, alkylphosphine, alkylphosphine oxide and other organphosphorus with wide usages, a conventional synthsis route by using phosphorus halide and phosphate to compound the organphosphorus is avoided, the synthesis steps are simple and short, the production technology is operated simply and easily, the reproducibility is good, the reaction is simple and green, the use of a phosphorus halide reagent is avoided, the range of the synthetic organphosphorus is wider, the method satisfies the requirements of green chemical development, and the large-scale production can be achieved.
Thermally induced structural transformations of linear coordination polymers based on aluminum tris(diorganophosphates)
D?bowski, Maciej,?okaj, Krzysztof,Ostrowski, Andrzej,Zachara, Janusz,Wiecińska, Paulina,Falkowski, Pawe?,Krztoń-Maziopa, Anna,Florjańczyk, Zbigniew
supporting information, p. 16480 - 16491 (2018/12/05)
The thermal transitions of inorganic-organic hybrid polymers composed of linear aluminum tris(diorganophosphate) chains with a general formula of catena-Al[O2P(OR)2]3 (where R = C1-C8 alkyl group or phenyl moiety) have been studied by means of DSC, powder XRD, TGA and TG-QMS, as well as optical spectroscopy. DSC and XRD reveal that most of them undergo reversible structural transformations in the solid state between ?100 and 200 °C caused by the changes in conformation of their organic substituents; however, a translational displacement of the rigid polymeric chains occurs only in the case of the derivative bearing long 2-ethylhexyl groups, which becomes liquid at about 140 °C. The thermal decomposition of the studied polymers begins between 200 and 265 °C depending on the type of organic substituent R decorating their aluminophospate core. TGA combined with mass spectrometry of the evolved gaseous products shows that the pyrolytic decomposition of Al[O2P(OR)2]3 proceeds either through β-elimination of olefin (for compounds with C2-C8 aliphatic ligands), or a homolytic cleavage of the P-OR bond (for methyl and phenyl derivatives); both processes are accompanied by condensation of the newly formed POH groups and liberation of water. Powder XRD, FTIR and SEM analyses of the solid residues indicate that thermolysis of Al[O2P(OR)2]3 accompanied by olefin elimination leads to the formation of condensed aluminum phosphates, mainly aluminum cyclohexaphosphate, exhibiting porous morphology. On the other hand, thermal degradation of methyl or phenyl derivatives results in amorphous aluminophosphate residues, and the latter contains conducting carbonaceous phases.